Biological Sciences
Health Sciences
Neuroscience
Understanding Amyotrophic Lateral Sclerosis (ALS) through TDP-43 Pathology


Alexis Park '28
Oct 31, 2025
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disease that affects the upper and lower motor neurons. The degeneration of the upper and lower motor neurons that send messages to the muscles causes them to weaken, fasciculate, and eventually undergo atrophy. As the connection between the brain and the muscles is damaged, the brain loses its ability to control voluntary movement, and symptoms of both upper and lower motor neuron damage coexist (Brotman 2024). Depending on the affected nerve cells, symptoms of ALS include weakness in the legs, feet, or ankles, as well as cognitive or behavioral changes. There is generally no pain in the early stages of ALS, and it often starts in the hands, feet, arms, or legs before spreading to other parts of the body as more nerve cells die (Mayo Clinic). Genetics, age, and sex are established risk factors for ALS, along with smoking and environmental toxin exposure, although the general etiology of ALS is unknown. With over 120 genes implicated in ALS, TDP-43 and FUS, genes for RNA-binding proteins, are prominently involved in familial ALS (Brotman 2024). TDP-43 aggregation is consequential to developing ALS, where clumps of misfolded or damaged proteins are found in cells and tissues (Housmans 2023).
Although TDP-43 is primarily localized to the nucleus, research suggests that TDP-43 in ALS mislocalizes to the cytoplasm and induces misfolding in other proteins due to its prion-like qualities. Evidence for prion-like propagation includes immunoblot analyses that found that C-terminal fragments of the TDP-43 acted as seeds that induced conformational changes across neural systems, and can help explain the progressive nature of ALS (Nonaka 2013). However, while misfolded TDP-43 does travel between cells through exosomes, microvesicles, or other extracellular vesicles similar to other prion diseases, TDP-43 proteinopathies are restricted to spreading within a single host, lacking the infectivity that characterizes the cross-host transmissibility of most prion diseases (Pongrácová 2024).
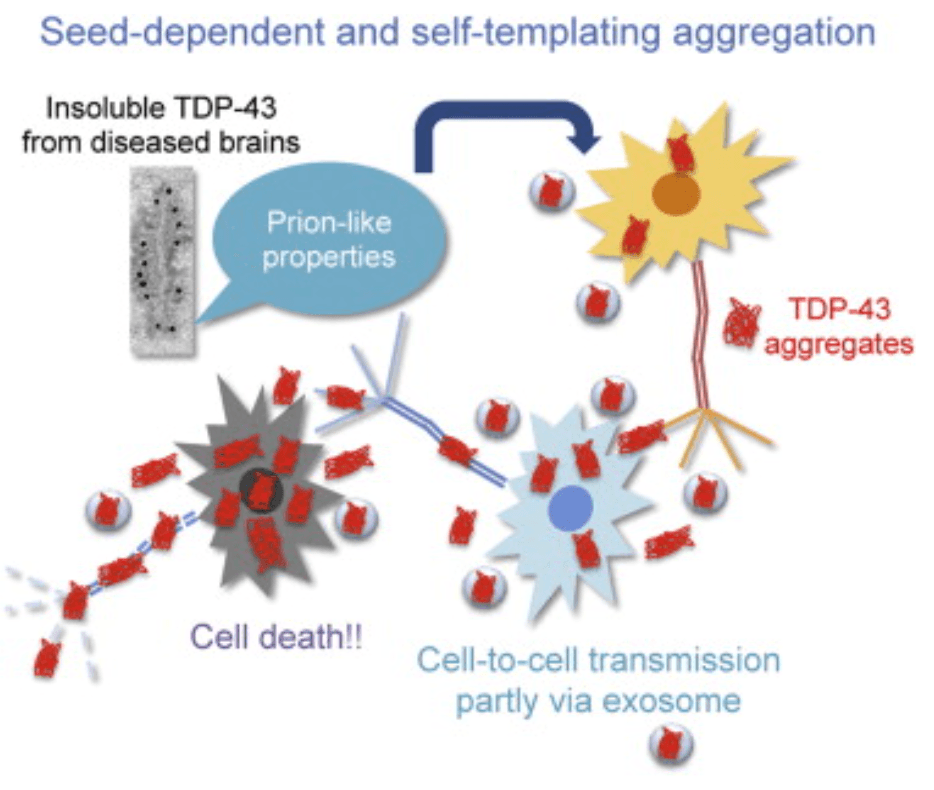
Figure 1: TDP-43 proteinopathy (Nonaka 2013)
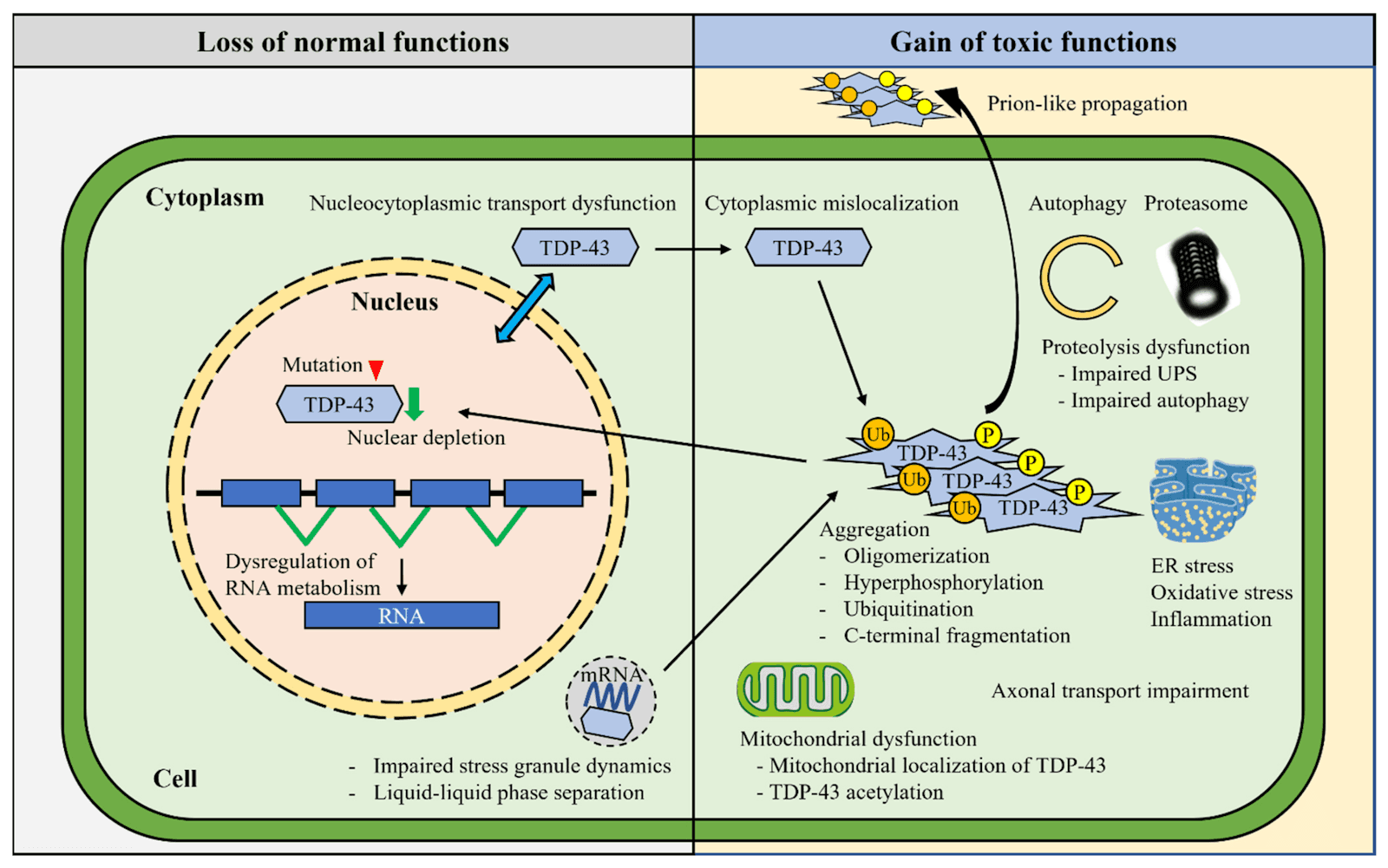
Figure 2: Molecular mechanisms underlying TDP-43 pathogenesis in the loss of normal functions in the nucleus and the gain of toxic functions in the cytoplasm (Tamaki 2022).
In healthy cells, TDP-43 constantly shuttles between the nucleus and the cytoplasm. This cycle allows for TDP-43 to perform RNA splicing and transcription in the nucleus, form ribonucleoprotein transport granules and stress granules in the cytoplasm, and return to the nucleus as needed. However, in pathological mislocalization, TDP-43 moves out of the nucleus and aggregates in the cytoplasm. This aggregation triggers post-translational modifications that lead to oligomerization, ubiquitination, hyperphosphorylation, and C-terminal fragmentation. As the aggregates trap more TDP-43, it becomes difficult for the TDP-43 to shuttle back to the nucleus, leading to a loss of normal functions such as RNA metabolism and gene expression regulation. The loss of function in the nucleus and cytoplasmic aggregation of TDP-43 lead to further cytotoxic effects such as impaired stress granule dynamics, liquid-liquid phase separation, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, axonal transport impairment, and proteolysis dysfunction that impairs autophagy and the Ubiquitin-Proteasome System (UPS) (Tamaki 2022). However, the mechanistic link from each feature to neurodegeneration in ALS is currently unknown, making it difficult to target specific features of TDP-43 dysfunction.
TDP-43 pathology is the most common downstream mechanism of ALS neurodegeneration, although absent when linked to the presence of SOD1 or FUS mutations (Lee 2012). Most ALS-associated TDP-43 mutations are dominant missense mutations in the glycine-rich domain (GRD) at the C-terminal. This domain plays both functional and structural roles, as the GRD is essential for facilitating protein-protein interactions and liquid-liquid phase separation that mediates the formation of membraneless organelles with essential cellular function (Carey 2022).
Ongoing efforts are focused on developing targeted therapies, such as Qalsody (Toferson), an intrathecal injection associated with SOD1 mutations. However, ALS diagnosis and treatment continue to lack disease-specific biomarkers. Diagnostic advancements are hampered by the extensive heterogeneity of ALS, along with difficulties in detecting pathological forms of TDP-43 in biofluids. This uncertainty is compounded by pathological observations. For example, it was found that in comparison to standard cases of sporadic ALS, patients who have had the disease for a longer duration (10-20 years) were noted to have lower TDP-43 aggregates. This suggests that neurons with TDP-43 aggregates are degenerate earlier on, further evidence in the spinal cord showing TDP-43 aggregates in the center of neuronophagic cell clusters, groups of immune cells that surround and digest dying neurons (Lee 2012). However, these findings do not resolve the critical question of whether TDP-43 aggregates themselves are toxic or are a marker for another neurotoxic process. Being able to validate either possibility is essential: if the aggregates are toxic, therapies could narrow their focus on preventing aggregate formation, clearance, and propagation; whereas, treatment would pivot to focusing on an upstream cause if the aggregates were proven to just be a marker (Suk 2020). Another significant concern is the translation of findings in model organisms, such as Yeast, C. elegans, and Drosophila, to human patients. For instance, a limitation of model organisms includes their inability to mirror the biological complexity of human ALS. Human ALS involves a complexity of genetic factors and an extensive nervous system that cannot be fully replicated, and new data have begun to show that cytoplasmic TDP-43 inclusions can adopt different protein folds in different human diseases (Armas 2025). However, based on the success of Qalsody, which is an RNA-based therapeutic, recent efforts have aimed to apply similar strategies to TDP-43. For example, Tamoxifen, an estrogen modulator used to treat breast cancer, was identified as a potential ALS treatment due to its ability to enhance autophagy. While it was hypothesized that Tamoxifen could therefore clear TDP-43 aggregates, a 2020 trial comparing patients assigned either Tamoxifen or a placebo showed a statistically insignificant difference between the two (Chen 2020). Ongoing therapies act as a stress granule modulator, a kinase inhibitor, and more.
TDP-43 proteinopathy has also presented typical symptoms of Parkinson’s disease, as cytoplasmic aggregation led to dopaminergic neuronal loss and motor symptoms (Yamashita 2022), and Alzheimer’s disease, aggregates most often distributed throughout the limbic system (Meneses 2021). Therefore, being able to appropriately understand and model TDP-43 proteinopathy is necessary, not only toward ALS, but a range of neurodegenerative diseases. However, recent technological advances in CRISPR-based methods may provide the necessary biological insight to pare down on the infinite array of possible treatments (Kampmann 2018).
Edited by Taanvi Gowdar ‘28
Sources
Armas, J.M.B., Taoro-González, L., Fisher, E.M.C. et al. “Challenges of modelling TDP-43 pathology in mice.” Mamm Genome 36, 465–481 (2025). https://doi.org/10.1007/s00335-025-10131-1
“Amyotrophic lateral sclerosis (ALS) - Symptoms and causes.” Mayo Clinic, https://www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms-causes/syc-20354022. Accessed 25 August 2025.
Brotman, Ryan. “Amyotrophic Lateral Sclerosis - StatPearls.” NCBI, https://www.ncbi.nlm.nih.gov/books/NBK556151/. Accessed 25 August 2025.
Carey, Jenny L., and Lin Guo. “Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases.” Frontiers in Molecular Biosciences vol. 9 826719. 2 Feb. 2022, doi:10.3389/fmolb.2022.826719
Chen, Po-Chih et al. “Tamoxifen for amyotrophic lateral sclerosis: A randomized double-blind clinical trial.” Medicine vol. 99,22 (2020): e20423. doi:10.1097/MD.0000000000020423
Housmans, Joëlle A J et al. “A guide to studying protein aggregation.” The FEBS journal vol. 290,3 (2023): 554-583. doi:10.1111/febs.16312
Kampmann, Martin. “A CRISPR Approach to Neurodegenerative Diseases.” Trends in Molecular Medicine vol. 23,6 (2017): 483-485. doi:10.1016/j.molmed.2017.04.003
Lee, Edward B et al. “Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration.” Nature Reviews Neuroscience vol. 13,1 38-50. 30 Nov. 2011, doi:10.1038/nrn3121
Meneses, Axel, et al. “TDP-43 Pathology in Alzheimer's Disease - Molecular Neurodegeneration.” Molecular Neurodegeneration, 20 December 2021, https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-021-00503-x. Accessed 25 August 2025.
Nonaka, Takashi, and Masami Masuda-Suzukake. “Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains.” Cell Reports, 11 July 2013, https://www.cell.com/cell-reports/fulltext/S2211-1247(13)00285-4.
Pongrácová, Emma, and Emanuele Buratti. “Prion-like Spreading of Disease in TDP-43 Proteinopathies.” MDPI, 25 October 2024, https://doi.org/10.3390/brainsci14111132.
Suk, Terry R., and Maxime W. C. Rousseaux. “The role of TDP-43 mislocalization in amyotrophic lateral sclerosis.” Molecular Neurodegeneration vol. 15,1 45. 15 Aug. 2020, doi:10.1186/s13024-020-00397-1
Tamaki, Yoshitaka. “Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD.” MDPI, 2022, https://www.mdpi.com/1422-0067/23/20/12508. Accessed 25 August 2025.
Yamashita, Rika, and Goichi Beck. “TDP-43 Proteinopathy Presenting with Typical Symptoms of Parkinson's Disease.” International Parkinson and Movement Disorder Society, 09 May 2022, https://movementdisorders.onlinelibrary.wiley.com/doi/10.1002/mds.29048.
Read More

Kevin He '29
Apr 4, 2026
Universal Base-Edited CAR7 T-Cell Therapy to Treat Relapsed T-cell Acute Lymphoblastic Leukemia (T-ALL)
Base-edited CAR-T therapy offers new hope for relapsed T-ALL by inducing remission and bridging patients to stem cell transplant.

Keigo Fujita '29
Apr 3, 2026
Rejuvenating Senescent Dopaminergic Neurons: A Novel Therapeutic Approach to Parkinson's Disease
A novel therapeutic strategy for Parkinson's Disease may soon restore neuronal function without neuron elimination or replacement.

Nina Prakash
Mar 9, 2026
Nina Prakash's Senior Thesis Spotlight: Investigating the role of the serotonin 1B receptor in the neuroplastic effects of psilocybin in mice
Nina Prakash ’25 studies whether psilocybin’s neuroplastic effects depend on serotonin 1B rather than serotonin 2A.